Das sogenannte Hutchinson-Gilford-Progerie-Syndrom ist zwar selten, aber dafür besonders grausam: Während andere Kinder wachsen und reifen, leiden die Betroffenen unter Wachstumsstörungen und verwandeln sich in junge Greise. Bei etwa einem von vier Millionen Kindern stellen Ärzte die entsprechenden Symptome oft schon im ersten oder zweiten Lebensjahr fest. Die Haut der Kinder beginnt zu altern, die Haare fallen aus und es entwickeln sich zunehmend Gelenkprobleme und Herz-Kreislauf-Erkrankungen. Die Betroffenen erreichten deshalb bisher oft nur ein Alter von etwa 14 Jahren. Im vergangenen Jahr gab es zwar bereits eine erfreuliche Nachricht für Progerie-Patienten: Das Medikament Lonafarnib wurde zugelassen, das eine gewisse Lebensverlängerung ermöglicht. Der Wirkstoff greift allerdings nur in einen bestimmten Ablauf im Rahmen der Krankheitsentstehung ein und ist dadurch nur begrenzt wirksam. Der neue Ansatz packt das Problem nun hingegen an der genetischen Wurzel.
Eine kleine Mutation mit fatalen Folgen
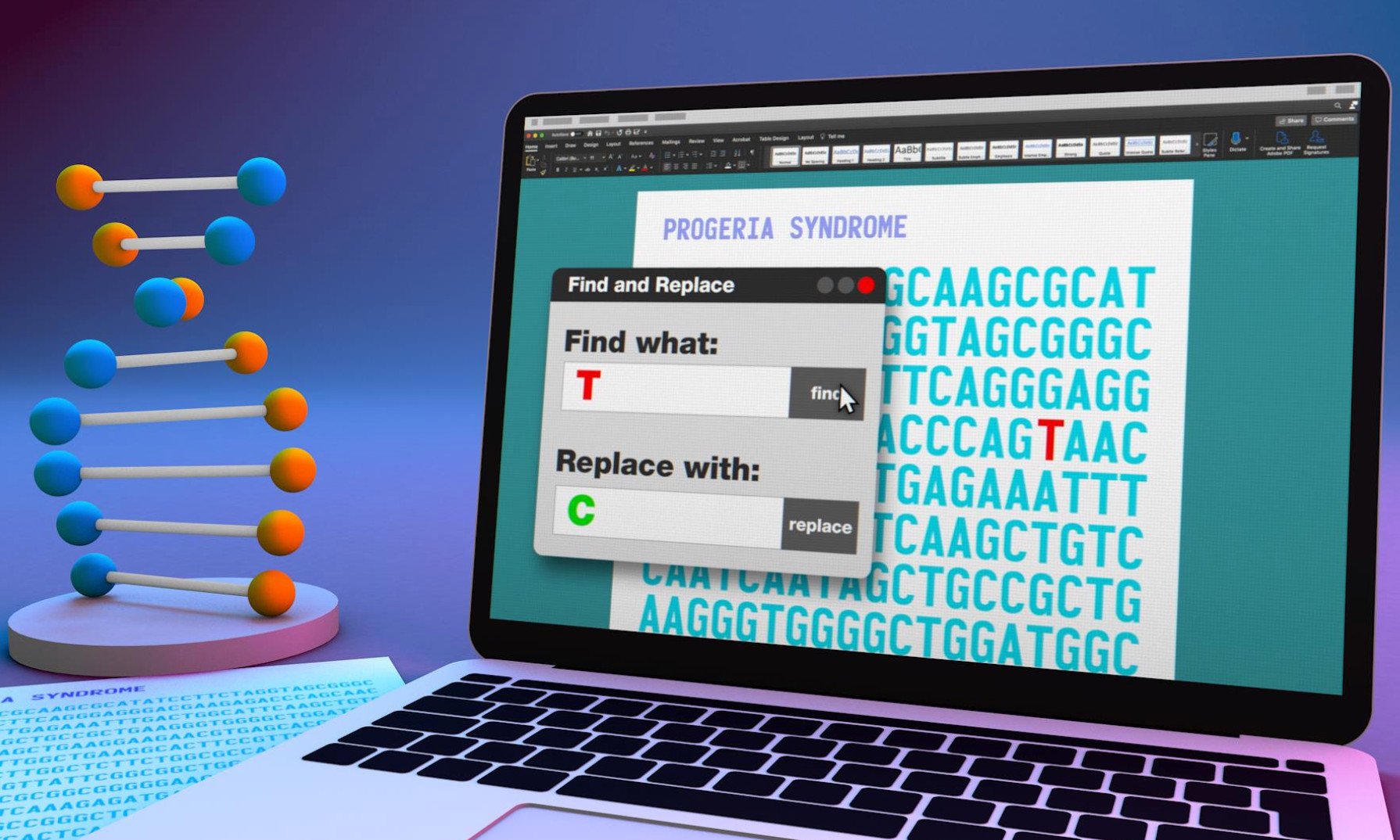
Die Ursache der Krankheit ist eine Mutation im sogenannten Lamin-A-Gen, das die Informationen für einen wichtigen Baustein der Kernmembran in den Zellen des Körpers trägt. Diese Erbanlage weist bei den Progerie-Patienten einen kleinen, aber schwerwiegenden Fehler auf: Bei ihrem Lamin-A-Gen ist eine DNA-Base von Cytosin (C) zu Thymin (T) verändert. Diese Mutation führt zur Produktion des Proteins Progerin, das den schnellen Alterungsprozess verursacht. An dieser Punktmutation hat nun ein Team von US-Forschern den Hebel angesetzt. “Die Tatsache, dass eine einzige spezifische Mutation die Krankheit verursacht, ermöglicht den Einsatz von DNA-Editing-Techniken zur Korrektur“, sagt Co-Autor Francis Collins vom National Human Genome Research Institute in Bethesda.
Es gab bereits Versuche, die bekannte Gen-Editing-Methode Crispr/Cas9 zu verwenden, um die Mutation in dem Gen gezielt zu reparieren. Doch dieses Verfahren birgt die Gefahr von Kollateralschäden: Es können problematische DNA-Bruchstücke entstehen oder Schnitte außerhalb des eigentlichen Zielbereiches. Die Forscher setzten deshalb nun die neuere Technik des DNA-Base-Editings ein. Dabei kommt eine abgewandelte Form des Crispr/Cas9-Systems zum Einsatz, durch die einzelne Basen an einer bestimmten genomischen Stelle verändert werden können. Der wichtige Unterschied ist dabei, dass die Basen-Editoren dabei nicht das Phosphatgerüst der DNA spalten, sodass keine doppelsträngigen DNA-Brüche entstehen können.
Vielversprechender Erfolg
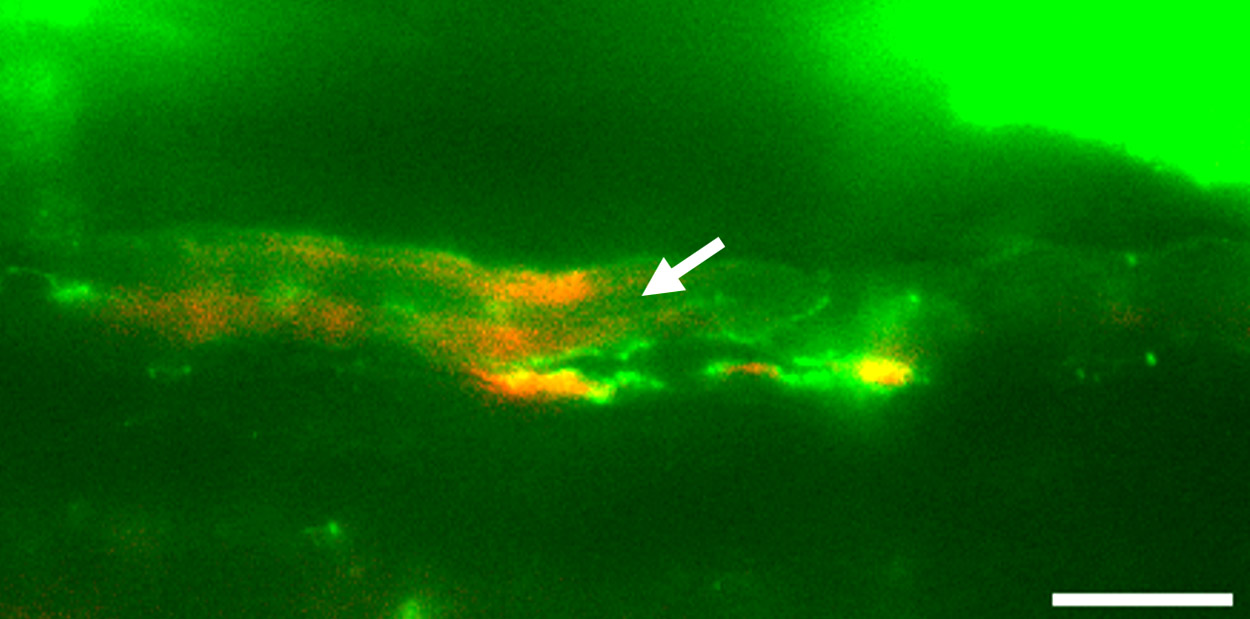
Die Forscher testeten die Base-Editing-Methode zur Korrektur des Gendefekts zunächst an im Labor gezüchteten Bindegewebszellen von Progerie-Patienten. Dies führte zu einem vielversprechenden Ergebnis: Die Behandlung konnte die Mutation in etwa 90 Prozent der Zellen korrigieren. So gingen die Forscher anschließend zu Tierversuchen über: Sie verwendeten dabei Mäusezuchtlinien, die ein Modell der Progerie darstellen. Die Tiere besitzen ebenfalls die entsprechende Mutation in dem Lamin-A-Gen, zeigen vorzeitige Alterungserscheinungen und besitzen eine deutlich verkürzte Lebenserwartung. Diesen „Progerie-Mäusen“ verabreichten die Wissenschaftler kurz nach der Geburt eine intravenöse Injektion der DNA-Editing-Substanz. Anschließend untersuchten sie den Effekt und die Entwicklung der Versuchstiere.